Module View is the standard program interface for the analysis of single molecule calculation results inherited from previous versions of ACD/Percepta.
After starting the program, paste a 2-D structure to the Structure Pane by clicking the button, or find and select a structure from the Dictionary by clicking the button. A calculation will be performed for the presented structure automatically.
Workspace Panel Switch between modules by clicking the appropriate item in the tree view. Click "+" and "-" nodes to expand/collapse the corresponding module group. The first time you select a module, algorithms are loaded to your PC’s operating memory. Depending on the speed of your PC, this can take up to one minute. All subsequent calculations take only one or two seconds.
Navigation Bar The Navigation Bar appears in all workspaces except the Spreadsheet View. It displays presented structure and two previous as well as two next stuctures in the corresponding dataset. If no dataset is selected, the Navigation Bar displays the history of performed calculations. When the compound outside of the dataset is used as an input structure for calculation, the user can add it to the current dataset: a. Click the pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound. b. Click the pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically. c. The structure browse bar displays the active structure number and the total number of records in the current dataset. Scroll through the database using the browse buttons: First/Last record, Previous/Next record. Type a record number to view it directly.
Structure Pane The Structure Pane displays a 2-D representation of the chemical structure used for calculation. A compound name is displayed only for those structures which are found in the Dictionary. (Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure. (Ctrl+C): Copies the active structure in the Structure Pane to the clipboard. (Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically. (Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+I): Type or paste InChI (International Chemical Identifier) notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats. (Ctrl+E): Allows to edit the active molecule.
Results Pane As its name implies this pane displays the results of calculations and predictions.
Saves a PDF report for each compound in the active project in the directory of your choice. Creates a PDF report with the displayed results. Copies the displayed results to the clipboard.
These icons appear only in those modules which involve trainable models. Opens the window to select the calculation algorithms (in LogD and LogS modules) or switch between the training libraries used in calculations. The name of the currently selected library is indicated with italic font. Allows to add new compounds to the editable training library. If the library is read-only, this button is inactive.
Similar Structures Pane This pane appears in all modules except BBB, pKa, LogD, LogS, Boiling point/Vapor pressure and Sigma modules, and displays up to the five most similar structures in the training set based on similarity criteria. If there are fewer than 5 or no structures displayed in this pane, the Tanimoto coefficient for the structure you have entered is less than 0.8 using MACCS-II keys. In simpler terms, this means that the structure may be outside the structural space covered by the training set that was used to build the algorithm. By hovering over any structure in this pane, a pop-up window with a larger representation of the structure and complete literature references will appear. In the Trainable Models similar structures are displayed according to their Similarity Indices (SI). If the compound that is being analyzed was in the training set, “Exact match” text will appear on such molecule in the Similar Structures Pane. Structures that represent compounds with lower Similarity Index values are shaded. Note: The calculated values for the input structure and the experimental one in the Similar Structures Pane may differ slightly, as the calculation for the input structure is performed by the algorithm, and the experimental values, displayed in the Similar Structures Pane, are taken from the references.
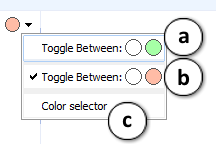
Click a circle to apply a label to the current compound. To set the color of the label, click the arrow button to expand the color settings list:
a. When clicking the label circle while this option is selected, the color of the label for the displayed compound will alternate between green or none.
b. When clicking the label circle while this option is selected, the color of the label for the displayed compound will alternate between red or none.
c. To set an arbitrary label color, select the "Color selector" option and click the label circle. The color palette will appear. Choose one color of your preference from a predefined set of colors.

 pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.
pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.  pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.
pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.  (Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure.
(Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure. (Ctrl+C): Copies the active structure in the Structure Pane to the clipboard.
(Ctrl+C): Copies the active structure in the Structure Pane to the clipboard. (Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically.
(Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically. (Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure.
(Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+I): Type or paste InChI (International Chemical Identifier) notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure.
(Ctrl+I): Type or paste InChI (International Chemical Identifier) notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats.
(Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats. (Ctrl+E): Allows to edit the active molecule.
(Ctrl+E): Allows to edit the active molecule.  Saves a PDF report for each compound in the active project in the directory of your choice.
Saves a PDF report for each compound in the active project in the directory of your choice. Creates a PDF report with the displayed results.
Creates a PDF report with the displayed results. Copies the displayed results to the clipboard.
Copies the displayed results to the clipboard. Opens the window to select the calculation algorithms (in LogD and LogS modules) or switch between the training libraries used in calculations. The name of the currently selected library is indicated with italic font.
Opens the window to select the calculation algorithms (in LogD and LogS modules) or switch between the training libraries used in calculations. The name of the currently selected library is indicated with italic font. Allows to add new compounds to the editable training library. If the library is read-only, this button is inactive.
Allows to add new compounds to the editable training library. If the library is read-only, this button is inactive.