Oral Bioavailability: Difference between revisions
No edit summary |
|||
| Line 61: | Line 61: | ||
<div class="mw-collapsible-content"> | <div class="mw-collapsible-content"> | ||
''' | ===Bioavailability DB=== | ||
'''Number of compounds:''' 788<br> | |||
'''Main sources of experimental data:''' | '''Main sources of experimental data:''' | ||
* Reference books: | * Reference books: | ||
| Line 79: | Line 75: | ||
<br> | <br> | ||
===Simulation model=== | |||
The mathematical model that is used for simulations performed by PK Explorer and Oral Bioavailability modules is based on differential equations describing solubility in the gastrointestinal tract, passive-absorption in jejunum and elimination (total body clearance). Other pharmacokinetic properties such as first-pass effect in liver and gut and volume of distribution are also considered in simulations. | |||
Quantitative %F values are calculated as a ratio of AUCs after oral and intravenous administration. Also, the employed simulation model allows evaluating the dose dependence of bioavailability. | |||
<br> | |||
===Validation=== | |||
====Validation Set & Assessment Procedure==== | |||
Since the predictions are based on a mechanistic simulation model rather that formal statistical fitting to a set of data points, the validation procedure was not based on a conventional training/test set approach. Instead, a set of clinical fraction absorbed (''f<sub>a</sub>'') data together with dosage information for 28 drugs reported by Parrott & Lavé [1] (originating mainly from a compilation by Zhao et al. [2]) was used for validation purposes. | |||
The model performance was assessed in several ways: | |||
* Qualitatively, by evaluating the accuracy of three-class classification, where the compounds were categorized by their calculated extent of absorption in the following manner: | |||
** Low: ''f<sub>a</sub>'' ≤ 33% | |||
** Moderate: 33% < ''f<sub>a</sub>'' < 66% | |||
** High: ''f<sub>a</sub>'' ≥ 66% | |||
** Quantitatively, using the Residual Mean Square Error (RMSE) statistic and visual inspection of the correlation between observed and predicted ''f<sub>a</sub>'' values of the considered drugs. | |||
==== | ====Validation results==== | ||
<br> | <br> | ||
<!-- TABLE 1 --> | <!-- TABLE 1 --> | ||
| Line 159: | Line 174: | ||
|} | |} | ||
</div> | </div> | ||
</div> | </div> | ||
Revision as of 13:39, 17 July 2013
Overview
Oral Bioavailability module predicts the fraction of the specified drug dose that reaches systemic circulation after oral administration (%F). For calculation of quantitative %F values, Oral bioavailability module uses the same kind of absorption simulation that is implemented in ACD/PK Explorer.
Features
- Predicts %F after oral administration with the possibility to explore dose-dependence of bioavailability
- Predicts a number of endpoints that affect oral bioavailability:
- Solubility (dose/solubility ratio)
- Stability in acidic media
- Intestinal membrane permeability by passive or active transport
- Likelihood of P-gp efflux
- Likelihood of first pass metabolism in the liver
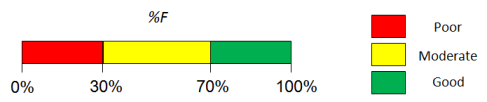
- Visualizes the contributions of underlying properties with traffic-lights (green = good, red = problematic) for easy interpretation
- Displays experimental %F values for up to 5 similar structures from Bioavailability DB along with literature references.
Interface

- 50 mg is the default oral drug dose used in calculations. Enter any desired value to explore the effect of dose on oral bioavailability.

- a. Click the "Undo" button to reset the specified drug dose and to recalculate %F using the default settings.
- b. Click to recalculate %F using the currently specified dose.
- Predicted %F (Oral) value.
- Click to see more details regarding the calculation of the particular property.
- Factors affecting oral bioavailability (see below for details).
- Hover over a title to view a screentip with a short description.
- Up to 5 similar structures in the Bioavailability DB with experimental values and references.


Traffic-lights system explanation
- Solubility in gastrointestinal tract:
- Green – good – dose/solubility ratio < 1.
- Yellow – moderate – dose/solubility ratio between 1 and 10.
- Red – poor – dose/solubility ratio > 10.
- Stability – susceptibility to acid hydrolysis in stomach:
- Red – only assigned to highly reactive compounds that decompose in stomach very quickly. Red light means that F (oral) <10%, overriding any probabilistic prediction (i.e., do not pay attention to predicted probabilities in this particular case).
- Passive absorption – ability to cross human intestinal membrane by passive diffusion:
- Red – intestinal passive absorption <30%. %F (oral) is always less than passive absorption.
- Green – good (>70%) passive absorption across intestinal barrier. Passive absorption does not effect %F.
- First-pass metabolism – susceptibility to metabolic transformations catalyzed by enzymes in liver and intestine:
- Red – high probability that first-pass metabolism is >50%. In this case %F (oral) is likely to be dose dependent and not to exceed 40%.
- Green – compound probably does not undergo significant first-pass metabolism.
- P-gp efflux – susceptibility to backward transport through intestinal membrane:
- Red – compound is P-glycoprotein substrate. This effect is mostly important when compound is metabolized by CYP3A4
- Active transport – susceptibility to active transport through intestinal membrane:
- Green – compound is actively transported by PepT1, ASBT or other enzymes.
- Red light never appears, as this factor can only increase %F (oral).
Technical information
Bioavailability DB
Number of compounds: 788
Main sources of experimental data:
- Reference books:
- Therapeutic Drugs, Dolery, C., Ed. 2nd Edition, Churchill Livingstone, New York, NY, 1999
- Clarke's Isolation and Identification of Drugs, Moffat, A.C., Jackson, J.V., Moss, M.S., Widdop, B., Eds. 2nd Edition, The Pharmaceutical Press, London, 1986
- Various articles from peer-reviewed scientific journals*
* - Both articles reporting oral bioavailability models by other authors (i.e. with larger collections of experimental data per article) and dealing with detailed experimental pharmacokinetic characterization (i.e. usually only several compounds per article) were available for the training set construction.
Simulation model
The mathematical model that is used for simulations performed by PK Explorer and Oral Bioavailability modules is based on differential equations describing solubility in the gastrointestinal tract, passive-absorption in jejunum and elimination (total body clearance). Other pharmacokinetic properties such as first-pass effect in liver and gut and volume of distribution are also considered in simulations. Quantitative %F values are calculated as a ratio of AUCs after oral and intravenous administration. Also, the employed simulation model allows evaluating the dose dependence of bioavailability.
Validation
Validation Set & Assessment Procedure
Since the predictions are based on a mechanistic simulation model rather that formal statistical fitting to a set of data points, the validation procedure was not based on a conventional training/test set approach. Instead, a set of clinical fraction absorbed (fa) data together with dosage information for 28 drugs reported by Parrott & Lavé [1] (originating mainly from a compilation by Zhao et al. [2]) was used for validation purposes.
The model performance was assessed in several ways:
- Qualitatively, by evaluating the accuracy of three-class classification, where the compounds were categorized by their calculated extent of absorption in the following manner:
- Low: fa ≤ 33%
- Moderate: 33% < fa < 66%
- High: fa ≥ 66%
- Quantitatively, using the Residual Mean Square Error (RMSE) statistic and visual inspection of the correlation between observed and predicted fa values of the considered drugs.
Validation results
| Subset | Observed | Calculated probability (p) | |||
|---|---|---|---|---|---|
| >0.5 | <0.5 | ||||
| Entire training set N = 788* |
True | 415 (52.7%) |
114 (14.5%) | ||
| False | 81 (10.3%) |
178 (22.6%) | |||
| Accuracy |
| ||||
| Sensitivity |
| ||||
| Specificity |
| ||||
* - Since ACD/Bioavailability model does not utilize GALAS modeling methodology, no additional parameters (i.e., RI) are available to enable additional filtering of more reliable predictions.
| Subset | Observed | Calculated probability (p) | |||
|---|---|---|---|---|---|
| >0.5 | <0.5 | ||||
| Entire training set N = 788* |
True | 172 (21.8%) |
126 (16.0%) | ||
| False | 44 (5.6%) |
446 (56.6%) | |||
| Accuracy |
| ||||
| Sensitivity |
| ||||
| Specificity |
| ||||