Created page with "'''PhysChem Profiler''' is a special workspace that integrates the output of all physicochemical property predictors included in '''ACD/Percepta''' and presents it a single st..."
'''PhysChem Profiler''' is a special workspace that integrates the output of all physicochemical property predictors included in '''ACD/Percepta''' and presents it a single straihgtforward interface.
'''PhysChem Profiler''' is a special workspace that integrates the output of all physicochemical property predictors included in '''ACD/Percepta''' and presents it a single straihgtforward interface.<br>
<br>
<!--A key requirement for compound ranking during test prioritization is the ability to discriminate between chemical structure candidates using reliable predictions of physicochemical and ADME properties, enabling foresight into potential safety concerns. ACD/Labs prediction software presents full-featured profiling capabilities that allow classifying suggested analogs as “Good” or “Bad” based on their calculated values. Dedicated modules are also available for most of these properties, enabling the researcher to obtain more information about the calculation of a particular parameter.
<!--A key requirement for compound ranking during test prioritization is the ability to discriminate between chemical structure candidates using reliable predictions of physicochemical and ADME properties, enabling foresight into potential safety concerns. ACD/Labs prediction software presents full-featured profiling capabilities that allow classifying suggested analogs as “Good” or “Bad” based on their calculated values. Dedicated modules are also available for most of these properties, enabling the researcher to obtain more information about the calculation of a particular parameter.
<br><br>
<br><br>
Line 15:
Line 16:
-->
-->
[[File:physchem_profiler.png]]
[[File:physchem_profiler.png]]<br>
<br>
<ol>
<ol>
<li>'''Navigation Bar'''<br> The '''Navigation Bar''' appears in all workspaces except the '''Spreadsheet View'''. It displays presented structure and two previous as well as two next stuctures in the corresponding dataset. If no dataset is selected, the '''Navigation Bar''' displays the history of performed calculations. <br> When the compound outside of the dataset is used as an input structure for calculation, the user can add it to the current dataset:<br> [[File:Navigation_bar.png|center]]<br>
<li>'''Navigation Bar'''<br> The '''Navigation Bar''' appears in all workspaces except the '''Spreadsheet View'''. It displays presented structure and two previous as well as two next stuctures in the corresponding dataset. If no dataset is selected, the '''Navigation Bar''' displays the history of performed calculations. <br> When the compound outside of the dataset is used as an input structure for calculation, the user can add it to the current dataset:<br><br> [[File:Navigation_bar.png|center]]<br>
<ol style="list-style-type:lower-latin">
<ol style="list-style-type:lower-latin">
<li>Click the [[File:History_confirm_icon.png]] pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound. </li>
<li>Click the [[File:History_confirm_icon.png]] pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound. </li>
Line 33:
Line 35:
<li>Ionizable groups are highlighted using color shading (red for acid, blue for base, purple for amphoteric). More intensive shading denotes strongest acid and base groups</li><br>
<li>Ionizable groups are highlighted using color shading (red for acid, blue for base, purple for amphoteric). More intensive shading denotes strongest acid and base groups</li><br>
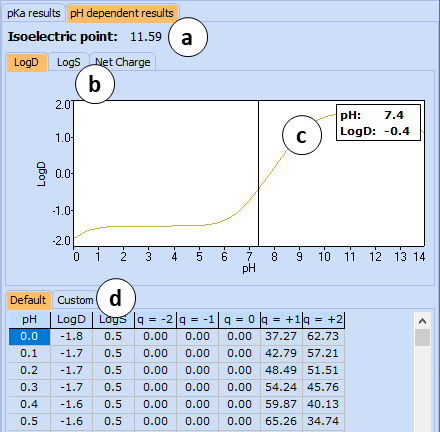
<li>The '''pH dependent results''' roll-down pane presents predicted values of ionization-dependent properties, such as octanol-water distribution (LogD) and solubility in buffer (LogS) in both graphical and tabular form.<br>
<li>[[File:physchem_profiler_phdependent_pane.png|right]]The '''pH dependent results''' roll-down pane presents predicted values of ionization-dependent properties, such as octanol-water distribution (LogD) and solubility in buffer (LogS) in both graphical and tabular form.<br><br>
<ol style="list-style-type:lower-latin">
<ol style="list-style-type:lower-latin">
<li>Click the tab of interest to view the plot of pH dependency of LogD, LogS, or Net Charge.[[File:physchem_profiler_phdependent_pane.png|right]]</li>
<li>Click the tab of interest to view the plot of pH dependency of LogD, LogS, or Net Charge.</li>
<li>Click and drag the slider to see the calculated logD values at precise pH value.</li>
<li>Click and drag the slider to see the calculated logD values at precise pH value.</li>
<li>The two tabs in the bottom part of this pane enable switching between ''Default'' and ''Custom'' views of the results table. The ''Default'' table is read-only and list calculated LogD, LogS, and fractions of the compound having different net charges throughout the entire pH scale (0-14) at 0.1 intervals. ''Custom'' table is editable and allows adding and deleting user-defined pH points.<br> <br>'''Note:''' Roll-down menus can be viewed simultaneously by dragging them partway up or down the pane.</li>
<li>The two tabs in the bottom part of this pane enable switching between ''Default'' and ''Custom'' views of the results table. The ''Default'' table is read-only and list calculated LogD, LogS, and fractions of the compound having different net charges throughout the entire pH scale (0-14) at 0.1 intervals. ''Custom'' table is editable and allows adding and deleting user-defined pH points.</li></ol><br> <br>'''Note:''' Roll-down menus can be viewed simultaneously by dragging them partway up or down the pane.</li><br>
</ol><br>
<li>A list of basic physicochemical and molecular properties:
<li>A list of basic physicochemical and molecular properties:
Revision as of 14:26, 21 February 2013
PhysChem Profiler is a special workspace that integrates the output of all physicochemical property predictors included in ACD/Percepta and presents it a single straihgtforward interface.
Navigation Bar The Navigation Bar appears in all workspaces except the Spreadsheet View. It displays presented structure and two previous as well as two next stuctures in the corresponding dataset. If no dataset is selected, the Navigation Bar displays the history of performed calculations. When the compound outside of the dataset is used as an input structure for calculation, the user can add it to the current dataset:
Click the pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.
Click the pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.
The structure browse bar displays the active structure number and the total number of records in the current dataset. Scroll through the database using the browse buttons: First/Last record, Previous/Next record or pressing the up/down arrow keys on the keyboard to go to the next/previous record. Type a record number to view it directly.
Structure Pane The Structure Pane displays a 2-D representation of the chemical structure used for calculation. A compound name is displayed only for those structures which are found in the Dictionary. (Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure. (Ctrl+C): Copies the active structure in the Structure Pane to the clipboard. (Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically. (Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats. (Ctrl+E): Allows to edit the active molecule.
Creates a PDF report with the displayed results. Copies the displayed results to the clipboard.
The pKa results roll-down pane displays the output of pKa predictions: strongest acid and base pKa values including reliability range in ±log units, a list of pKa constants for all stages of ionization, as well as a list of partial ionization reactions (microstages) responsible for each ionization stage. Contribution of each microstage to the final pKa value is given in percent. The results may slightly differ depending on the designated preferences of the Expert View. For more information see How to set the default algorithms for logP, pKa and LogS calculation? and pKa
Ionizable groups are highlighted using color shading (red for acid, blue for base, purple for amphoteric). More intensive shading denotes strongest acid and base groups
The pH dependent results roll-down pane presents predicted values of ionization-dependent properties, such as octanol-water distribution (LogD) and solubility in buffer (LogS) in both graphical and tabular form.
Click the tab of interest to view the plot of pH dependency of LogD, LogS, or Net Charge.
Click and drag the slider to see the calculated logD values at precise pH value.
The two tabs in the bottom part of this pane enable switching between Default and Custom views of the results table. The Default table is read-only and list calculated LogD, LogS, and fractions of the compound having different net charges throughout the entire pH scale (0-14) at 0.1 intervals. Custom table is editable and allows adding and deleting user-defined pH points.
Note: Roll-down menus can be viewed simultaneously by dragging them partway up or down the pane.
A list of basic physicochemical and molecular properties:

 pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.
pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.  pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.
pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.  (Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure.
(Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure. (Ctrl+C): Copies the active structure in the Structure Pane to the clipboard.
(Ctrl+C): Copies the active structure in the Structure Pane to the clipboard. (Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically.
(Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically. (Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure.
(Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats.
(Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats. (Ctrl+E): Allows to edit the active molecule.
(Ctrl+E): Allows to edit the active molecule.  Creates a PDF report with the displayed results.
Creates a PDF report with the displayed results. Copies the displayed results to the clipboard.
Copies the displayed results to the clipboard.