'''PhysChem Profiler''' is a special workspace that integrates the output of all physicochemical property predictors included in '''ACD/Percepta''' and presents it a single straihgtforward interface.<br>
'''PhysChem Profiler''' is a special workspace that integrates the output of all physicochemical property predictors included in '''ACD/Percepta''' and presents it a single straihgtforward interface.<br>
<br>
<br>
Line 17:
Line 15:
</ul>
</ul>
-->
-->
==Interface==
[[File:physchem_profiler.png|center]]<br>
[[File:physchem_profiler.png|center]]<br>
Revision as of 09:50, 20 March 2013
PhysChem Profiler is a special workspace that integrates the output of all physicochemical property predictors included in ACD/Percepta and presents it a single straihgtforward interface.
Navigation Bar The Navigation Bar appears in all workspaces except the Spreadsheet View. It displays presented structure and two previous as well as two next stuctures in the corresponding dataset. If no dataset is selected, the Navigation Bar displays the history of performed calculations. When the compound outside of the dataset is used as an input structure for calculation, the user can add it to the current dataset:
Click the pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.
Click the pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.
The structure browse bar displays the active structure number and the total number of records in the current dataset. Scroll through the database using the browse buttons: First/Last record, Previous/Next record or pressing the up/down arrow keys on the keyboard to go to the next/previous record. Type a record number to view it directly.
Structure Pane The Structure Pane displays a 2-D representation of the chemical structure used for calculation. A compound name is displayed only for those structures which are found in the Dictionary. (Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure. (Ctrl+C): Copies the active structure in the Structure Pane to the clipboard. (Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically. (Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats. (Ctrl+E): Allows to edit the active molecule.
Creates a PDF report with the displayed results. Copies the displayed results to the clipboard.
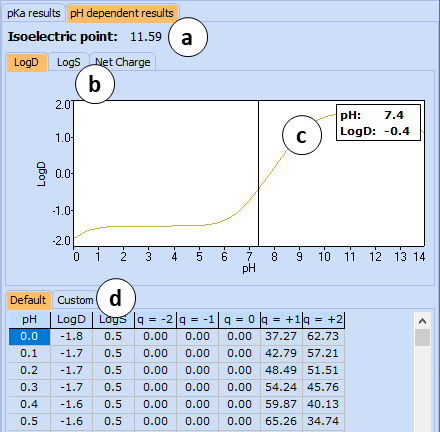
The pKa results roll-down pane displays the output of pKa predictions: strongest acid and base pKa values including reliability range in ±log units, a list of pKa constants for all stages of ionization, as well as a list of partial ionization reactions (microstages) responsible for each ionization stage. Contribution of each microstage to the final pKa value is given in percent. The results may slightly differ depending on the designated preferences of the Expert View. For more information see How to set the default algorithms for logP, pKa and LogS calculation? and pKa
Ionizable groups are highlighted using color shading (red for acid, blue for base, purple for amphoteric). More intensive shading denotes strongest acid and base groups
The pH dependent results roll-down pane presents predicted values of ionization-dependent properties, such as octanol-water distribution (LogD) and solubility in buffer (LogS) in both graphical and tabular form.
Click the tab of interest to view the plot of pH dependency of LogD, LogS, or Net Charge.
Click and drag the slider to see the calculated logD values at precise pH value.
The two tabs in the bottom part of this pane enable switching between Default and Custom views of the results table. The Default table is read-only and list calculated LogD, LogS, and fractions of the compound having different net charges throughout the entire pH scale (0-14) at 0.1 intervals. Custom table is editable and allows adding and deleting user-defined pH points.
Note: Roll-down menus can be viewed simultaneously by dragging them partway up or down the pane.
A list of basic physicochemical and molecular properties:
Algorithms for Calculating Basic Physicochemical Properties
At the heart of the additive-constitutive calculation algorithm of all physicochemical properties in ACD/Percepta lies the presumption that these properties can be estimated using additive atomic or group increments. Apart from molecular weight (MW), which is trivial to calculate, the algorithms may be divided into three general groups:
Derived macroscopic properties: density (d), refractive index (n), and surface tension (γ)
The dielectric constant ε (Permittivity)
Basic macroscopic properties such as molar volume (MV), molar refractivity (MR), and the parachor (Pr) are calculated first for the input structure. The atomic additive increments in such an algorithm depend on the bonds (single, double, aromatic, etc.) of this atom and on neighboring atoms. ACD/Percepta rapidly analyzes the input structure to determine the class of each atom, i.e., whether it is cyclic, aromatic, aliphatic, etc.
The prediction algorithms for density (d), refractive index (n) and surface tension (γ) are founded on well-known physicochemical formula which can be found in literature on physicochemical properties of compounds. They express d, n, and γ as functions of MV, MR, or Pr. Once the MV, MR, or Pr, have been predicted by additive means, it is straightforward to predict d, n, and γ using these formula.
The determination of the additive-constitutive atomic increments for MV, MR, and Pr were obtained internally by ACD/Labs scientists using large experimental databases relating structure to density, refractive index, and surface tension. The MV, MR, and Pr were recalculated from d, n, and γ. These parameters are proprietary information of ACD/Labs, Inc.
The prediction of the dielectric constant ε (permittivity) resembles very closely the prediction of boiling point, which is available as a separate Prediction Module in ACD/Percepta. Senior scientists at ACD/Labs discovered an additive function, which relates the dielectric constant to other macroscopic properties which can be additively treated, such as MV. Once this relationship was discovered, the additive-constitutive atomic increments for this function were obtained using large databases consisting of molecular structures and their observed dielectric constants. Using the function and estimated MV for the input structure, its dielectric constant can be quickly predicted.
Molar Volume, MV
By definition,
MV = MW / d
ACD/Percepta calculates molar volume from additive increments. The additive atomic increments were obtained using a database of density and calculated MW.
Note: The obtained MV value is used as an input parameter for further calculations, but is not displayed in the module interface itself.
Molar Refractivity, MR
The Lorentz-Lorenz equation relates refractive index, density, and refractive index:
MR = (n2 - 1) / (n2 + 2) x MW / d
ACD/Percepta calculates molar refractivity from additive increments. The additive atomic increments were obtained using a database of density, refractive index, and calculated MW.
Note: The obtained MR value is used as an input parameter for further calculations, but is not displayed in the module interface itself.
Parachor, Pr
By definition,
Pr = (MW / d) γ1/4
ACD/Percepta calculates the parachor from additive increments. The additive atomic increments were obtained using a database of density, surface tension, and calculated MW.
Density, d
By definition,
d = MW / MV
ACD/Percepta calculates the density from MW and the calculated molar volume (see above).
Refractive Index, n
By the Lorentz-Lorenz equation,
ACD/Percepta calculates the refractive index from the molar volume and molar refractivity, both of which are calculated as above.
Surface Tension, γ
By definition,
γ = (Pr / MV)4
ACD/Percepta calculates the surface tension from calculated MV (see above) and calculated Pr (see above).
Dielectric Constant, ε (Permittivity)
By definition,
f(ε) = f(MV, Additive Function)
ACD/Percepta calculates the dielectric constant from calculated MV (see above) and a proprietary empirical additive function.
Polarizability
This property is calculated from the Molar Refractivity (MR) as follows:
Polarizability = 0.3964308∙MR
Correlation Statistics with Experimental Data
Distribution of Molar Refractivity Prediction Error
Fast and reliable estimation of molecular transport properties is one of the key factors accelerating the process of drug discovery and development. Traditionally, calculated values of the octanol-water partition coefficient have been used for this purpose. In recent years, several new parameters have been introduced for absorption prediction, including molecular size and shape descriptors, hydrogen-bonding capabilities, and surface properties.
Another helpful parameter for the prediction of absorption is the polar surface area (PSA), defined as the sum of the surfaces of polar atoms in a molecule. PSA is a descriptor that was shown to correlate well with passive molecular transport through membranes, and therefore, allows the prediction of the transport properties of drugs. The calculation of PSA, however, used to be rather time consuming because of the necessity to generate a reasonable 3D molecular geometry and the calculation of the surface itself.
A new approach for the calculation of PSA is based on the summation of tabulated surface contributions of polar fragments. The method, termed topological PSA (TPSA), provides results that are practically identical to the 3D PSA, while the computation speed is 2–3 times of a magnitude faster.
Reference: Peter Ertl, Bernhard Rohde, and Paul Selzer. Fast calculation of Molecular Polar Surface Area as a Sum of Fragment-based contributions and its Application to the prediction of drug transport properties. J. Med. Chem.2000, 43, 3714–3717. [1]
The Bioconcentration Factor (BCF)
Bioconcentration is the increase in concentration of a chemical in an organism resulting from tissue absorption levels exceeding the rate of metabolism and excretion.
Bioconcentration Factor (BCF) is used to describe the accumulation of chemicals in organisms, primarily aquatic, that live in contaminated environments.
According to EPA guidelines, "the BCF is defined as the ratio of chemical concentration in the organism to that in surrounding water. Bioconcentration occurs through uptake and retention of a substance from water only, through gill membranes or other external body surfaces. In the context of setting exposure criteria, it is generally understood that the terms "BCF" and "steady-state BCF" are synonymous. A steady-state condition occurs when the organism is exposed to a sufficient length of time that the ratio does not change substantially."
Bioconcentration factors (BCFs) are used to relate pollutant residues in aquatic organisms to the pollutant concentration in ambient waters. The bioconcentration factor (BCF) is related to biomagnification effects. Many chemical compounds, especially those with a hydrophobic component, partition easily into the lipids and lipid membranes of organisms and bioaccumulate. If the compounds are not metabolized as fast as they are consumed, there can be significant magnification of potential toxicological effects up the food chain. The concern about bioaccumulation and biomagnification comes mainly from experience with chlorinated compounds, especially pesticides and PCBs, and their deleterious effects on vulnerable species, especially birds, frogs, and fish. Only minimal experimental and monitoring information has been gathered on the bioaccumulation properties of many other currently used chemical compounds. In fact, the biomagnification of many widely available chemicals has not been observed or predicted in aquatic systems.
BCF values are based on U.S. Environmental Protection Agency publications pursuant to Section 304(a) of the Federal Water Pollution Control Act as amended, literature values, or site-specific bioconcentration data. Current EPA guidelines for the derivation of human health water quality criteria use BCFs as well.
BCFs are quantified by various procedures depending on the lipid solubility of the pollutant. For lipid soluble pollutants, the average BCF is calculated from the weighted average percent lipids in the edible portions of fish and shellfish, about 3%; or it is calculated from theoretical considerations using the octanol-water partition coefficient. To estimate the BCF of a chemical compound, the quantitative structure activity relationship (QSAR) can be used:
logBCF = 0.79 x logPo/w - 0.40
where logP is the octanol-water partition coefficient. The "rule of thumb" is that a BCF value greater than 1000 poses a concern for bioaccumulation, particularly if the chemical is considered persistent in aquatic environments. This BCF value corresponds to a logP of 4.3 or greater.
For non-lipid soluble compounds, the BCF is determined empirically. The assumed water consumption is taken from the National Academy of Sciences publication, Drinking Water and Health (1977). (Referenced in the Human Health Guidelines.) This value, of 2.0 liters/day, is appropriate as it includes a margin of safety so that the general population is protected. The 6.5 grams per day of contaminated fish and shellfish consumption value is the average per-capita consumption rate of all (contaminated and non-contaminated) freshwater fish and shellfish for the U.S. population.
Although BCF assessments began as aquatic measurements, the exposure of plants and cattle to certain chemicals is also rated in terms of the bioconcentration factors.
Impact of BCFs on Humans
The human daily intake of chemical compounds is estimated assuming a "standard" daily intake of edible plants, meat, milk, and drinking water. The chemical accumulation in plants takes place both via the soil and the air. The BCFs are calculated as a function of logP only. The BCF for the plant accumulating chemicals via the air is estimated to be of minor importance for chemicals present in sludge. It is primarily estimated on the basis of the air-water partition coefficient of the chemical (which primarily depends on Henry’s constant) and logP.
The intake of chemicals by cattle, which may result in unacceptable concentrations in meat and milk, is characterized by the BCF calculated as a function of logP alone. In order to estimate the concentration levels in cattle and plants, the concentration of the chemicals in soil and interstitial water need to be known. Concentrations can be estimated on the basis of a general exposure model, and considerations for single chemicals are generalized for chemical mixtures assuming linear behavior. A "safe" environmental and hygiene risk assessment cannot be achieved by assessment only of a relative few of the many hundreds of chemical residues contaminating, waste water, soil, etc., so much effort has been put into developing an exposure assessment methodology sophisticated enough for chemical/physical well-defined fractions, which may be handled in accordance with procedures for single chemicals.
References and Examples of Use of BCF
Davis JW. Carpenter CL. “Environmental assessment of the alkanolamines.” Reviews of Environmental Contamination & Toxicology. 149:87–137, 1997.
Svendsen C. Weeks JM. “Relevance and applicability of a simple earthworm biomarker of copper exposure. II. Validation and applicability under field conditions in a mesocosm experiment with Lumbricus rubellus.” Ecotoxicology & Environmental Safety. 36(1):80–8, 1997 Feb.
Svendsen C. Weeks JM. “Relevance and applicability of a simple earthworm biomarker of copper exposure. I. Links to ecological effects in a laboratory study with Eisenia andrei.” Ecotoxicology & Environmental Safety. 36(1):72–9, 1997 Feb.
Falandysz J. Chwir A. “The concentrations and bioconcentration factors of mercury in mushrooms from the Mierzeja Wislana sand-bar, northern Poland.” Science of the Total Environment. 203(3):221–8, 1997 Sep 15.
Traas TP. Luttik R. Jongbloed RH. “A probabilistic model for deriving soil quality criteria based on secondary poisoning of top predators. I. Model description and uncertainty analysis.” Ecotoxicology & Environmental Safety. 34(3):264–78, 1996 Aug.
Wang X. Harada S. Watanabe M. Koshikawa H. Sato K. Kimura T. “Determination of bioconcentration potential of tetrachloroethylene in marine algae by 13C.” Chemosphere. 33(5):865–77, 1996 Sep.
Pant A. Srivastava SC. Singh SP. “Factors regulating methyl mercury uptake in a cyanobacterium.” Ecotoxicology & Environmental Safety. 32(1):87–92, 1995 Oct.
Makela TP. Oikari AO. “Pentachlorophenol accumulation in the freshwater mussels Anodonta anatina and Pseudanodonta complanata, and some physiological consequences of laboratory maintenance.” Chemosphere. 31(7):3651–62, 1995 Oct.
Brown DG. Lanno RP. van den Heuvel MR. Dixon DG. “HPLC determination of plasma thiocyanate concentrations in fish blood: application to laboratory pharmacokinetic and field-monitoring studies.” Ecotoxicology & Environmental Safety. 30(3):302–8, 1995 Apr.
Larry N. Britton (CONDEA Vista Company, Austin, Texas). Surfactants and the Environment. JSD 1,109–117 (1998).
Adsorption Coefficient (Koc)
The organic carbon adsorption coefficient, Koc, is crucial for estimating a chemical compound’s mobility in soil, and the prevalence of leaching from soil. The adsorption of a compound increases with an increase in organic content, clay content, and surface area of soil. The presence of a chemical compound can also be detected in groundwater, and inference can be made about its residence time in the soil and the degradation period before reaching the water table. The presence of continuous pores or channels in soil will increase the mobility of a chemical compound in the soil.
The Adsorption Coefficient (Koc) may be defined as the ratio of the amount of chemical adsorbed per unit weight of organic carbon (oc) in the soil or sediment to the concentration of the chemical in solution at equilibrium:
Koc = (μg adsorbed / g organic carbon) / (μg / mL solution)
Koc is calculated using the following equation:
logKoc = 0.544 x logPo/w + 1.377
References and Sample Uses of Koc
Vaussenat F. Bosc JY. LeBlanc M. Canaud B. “Data acquisition system for dialysis machines. A model for membrane hydraulic permeability.” ASAIO Journal. 43(6):910–5, 1997 Nov–Dec.
Jop KM. Guiney PD. Christensen KP. Silberhorn EM. “Environmental fate assessment of two synthetic polycarboxylate polymers.” Ecotoxicology & Environmental Safety. 37(3):229–37, 1997 Aug.
Heuman DM. Bajaj RS. Lin Q. “Adsorption of mixtures of bile salt taurine conjugates to lecithin-cholesterol membranes: implications for bile salt toxicity and cytoprotection.” Journal of Lipid Research. 37(3):562–73, 1996 Mar.
Underhill DW. “The adsorption of argon, krypton, and xenon on activated charcoal.” Health Physics. 71(2):160–6, 1996 Aug.
Scarpitta SC. “A new beaded carbon molecular sieve sorbent for 222Rn monitoring.” Health Physics. 70(5):673–9, 1996 May.
el-Kheshen S. Zia H. Badawi A. Needham TE. Luzzi LA. “Coating charcoal with polyacrylate-polymethacrylate copolymer for haemoperfusion. III: The effect of the coat thickness on the adsorption capacity of the coated charcoal and its adsorptivity to small and middle size molecules.” Journal of Microencapsulation. 12(5):505–14, 1995 Sep–Oct.
Scarpitta SC. “A theoretical model for 222Rn adsorption on activated charcoal canisters in humid air based on Polanyi's potential theory [see comments].” Comment in: Health Physics 1996 Feb;70(2):271–3 Health Physics. 68(3):332–9, 1995 Mar.

 pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.
pictogram to replace the current structure with the new one. All subsequent calculations will be performed automatically for the added compound.  pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.
pictogram to add new compound to the current dataset. It will appear as the last record in the dataset and all subsequent calculations will be performed automatically.  (Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure.
(Ctrl+V): Pastes an input structure from the clipboard to the Structure Pane. Also, if you are pasting a SMILES string from the clipboard, you can save a step by clicking directly on this button to display the 2-D structure. The calculation will be performed automatically for the input structure. (Ctrl+C): Copies the active structure in the Structure Pane to the clipboard.
(Ctrl+C): Copies the active structure in the Structure Pane to the clipboard. (Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically.
(Ctrl+D): Opens the Dictionary with >117,000 available names to find the corresponding structure and display it. When a compound is selected from the list and the selection confirmed by clicking OK, calculations for the chosen compound will be performed automatically. (Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure.
(Ctrl+S): Type or paste SMILES notation string in this field. The string will automatically be converted to 2-D structure. Calculation will be performed automatically for the input structure. (Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats.
(Ctrl+O): Opens a file containing a molecular structure. ACD/Percepta supports all versions of MDL *.mol files, ACD/ChemSketch *.sk2, as well as ISIS Draw *.skc, and ChemDraw *.cdx formats. (Ctrl+E): Allows to edit the active molecule.
(Ctrl+E): Allows to edit the active molecule.  Creates a PDF report with the displayed results.
Creates a PDF report with the displayed results. Copies the displayed results to the clipboard.
Copies the displayed results to the clipboard.